Variance partitioning analysis
Developed by Gabriel Hoffman
Run on 2026-03-19 09:17:05
Source:vignettes/variancePartition.Rmd
variancePartition.RmdIntroduction
Gene expression datasets are complicated and have multiple sources of
biological and technical variation. These datasets have recently become
more complex as it is now feasible to assay gene expression from the
same individual in multiple tissues or at multiple time points. The
variancePartition package implements a statistical method
to quantify the contribution of multiple sources of variation and
decouple within/between-individual variation. In addition,
variancePartition produces results at the gene-level to
identity genes that follow or deviate from the genome-wide trend.
The variancePartition package provides a general
framework for understanding drivers of variation in gene expression in
experiments with complex designs. A typical application would consider a
dataset of gene expression from individuals sampled in multiple tissues
or multiple time points where the goal is to understand variation within
versus between individuals and tissues. variancePartition
use a linear mixed model to partition the variance attributable to
multiple variables in the data. The analysis is built on top of the
lme4 package (Bates et al.
2015), and some basic knowledge about linear mixed models will
give you some intuition about the behavior of
variancePartition (Pinheiro and
Bates 2000; Galecki and Burzykowski 2013).
Input data
There are three components to an analysis:
- Gene expression data: In general, this is a matrix of normalized gene expression values with genes as rows and experiments as columns.
-
Count-based quantification:
featureCounts(Liao et al. 2014),HTSeq(Anders et al. 2015)Counts mapping to each gene can be normalized using counts per million (CPM), reads per kilobase per million (RPKM) or fragments per kilobase per million (FPKM). These count results can be processed with
limma::voom()(Law et al. 2014) to model the precision of each observation orDESeq2(Love et al. 2014). -
Isoform quantification:
kallisto(Bray et al. 2016),sailfish(Patro et al. 2014),salmon(Patro et al. 2015),RSEM(Li and Dewey 2011),cufflinks(Trapnell et al. 2010)These perform isoform-level quantification using reads that map to multiple transcripts. Quantification values can be read directly into R, or processed with
ballgown(Frazee et al. 2015) ortximport(Soneson et al. 2015). Microarray data: any standard normalization such as
rmain theoligo(Carvalho and Irizarry 2010) package can be used.Any set of features: chromatin accessibility, protein quantification, etc
-
Metadata about each experiment:
A
data.framewith information about each experiment such as patient ID, tissue, sex, disease state, time point, batch, etc. -
Formula indicating which metadata variables to consider:
An R formula such as
~ Age + (1|Individual) + (1|Tissue) + (1|Batch)indicating which metadata variables should be used in the analysis.
Variance partitioning analysis will assess the contribution of each metadata variable to variation in gene expression and can report the intra-class correlation for each variable.
Running an analysis
A typical analysis with variancePartition is only a few
lines of R code, assuming the expression data has already been
normalized. Normalization is a separate topic addressed briefly in
[Applying variancePartition to RNA-seq expression
data].
The simulated dataset included as an example contains measurements of
200 genes from 100 samples. These samples include assays from 3 tissues
across 25 individuals processed in 4 batches. The individuals range in
age from 36 to 73. A typical variancePartition analysis
will assess the contribution of each aspect of the study design
(i.e. individual, tissue, batch, age) to the expression variation of
each gene. The analysis will prioritize these axes of variation based on
a genome-wide summary and give results at the gene-level to identity
genes that follow or deviate from this genome-wide trend. The results
can be visualized using custom plots and can be used for downstream
analysis.
Standard application
# load library
library("variancePartition")
# load simulated data:
# geneExpr: matrix of gene expression values
# info: information/metadata about each sample
data(varPartData)
# Specify variables to consider
# Age is continuous so model it as a fixed effect
# Individual and Tissue are both categorical,
# so model them as random effects
# Note the syntax used to specify random effects
form <- ~ Age + (1 | Individual) + (1 | Tissue) + (1 | Batch)
# Fit model and extract results
# 1) fit linear mixed model on gene expression
# If categorical variables are specified,
# a linear mixed model is used
# If all variables are modeled as fixed effects,
# a linear model is used
# each entry in results is a regression model fit on a single gene
# 2) extract variance fractions from each model fit
# for each gene, returns fraction of variation attributable
# to each variable
# Interpretation: the variance explained by each variables
# after correcting for all other variables
# Note that geneExpr can either be a matrix,
# and EList output by voom() in the limma package,
# or an ExpressionSet
varPart <- fitExtractVarPartModel(geneExpr, form, info)
# sort variables (i.e. columns) by median fraction
# of variance explained
vp <- sortCols(varPart)
# Figure 1a
# Bar plot of variance fractions for the first 10 genes
plotPercentBars(vp[1:10, ])## Warning in geom_bar(stat = "identity", width = width): Ignoring empty aesthetic: `width`.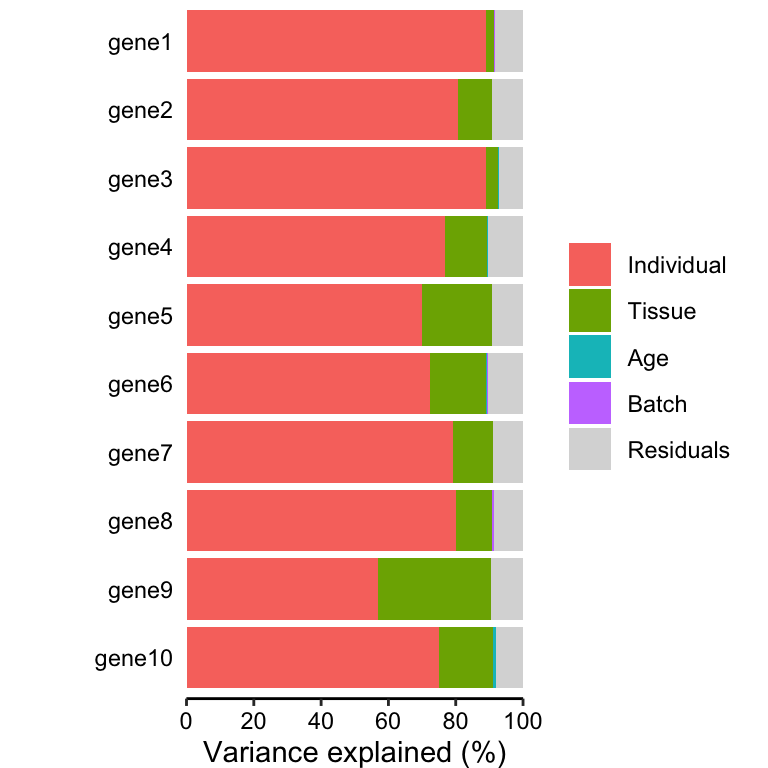
# Figure 1b
# violin plot of contribution of each variable to total variance
plotVarPart(vp)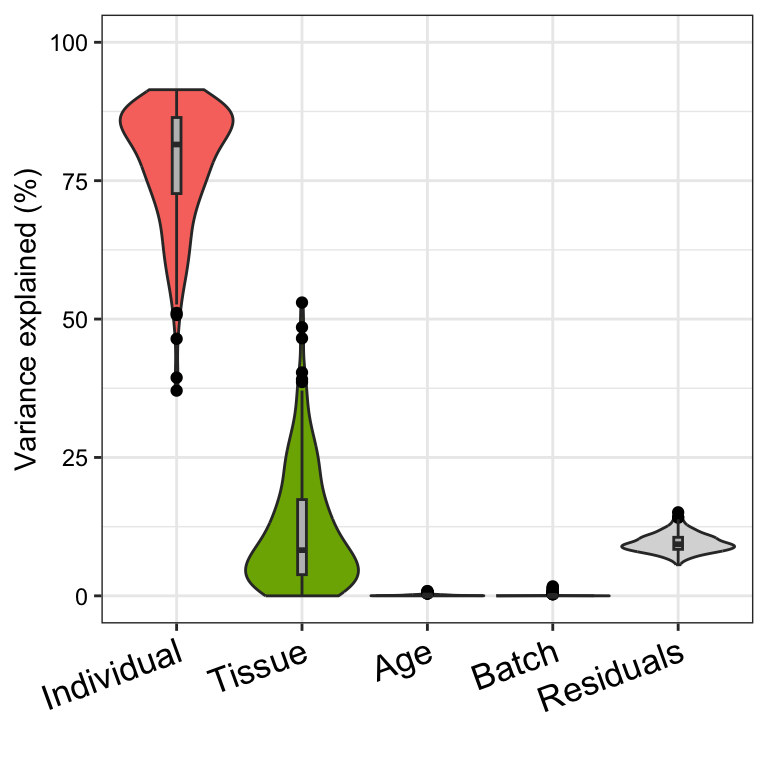
variancePartition includes a number of custom plots to
visualize the results. Since variancePartition attributes
the fraction of total variation attributable to each aspect of the study
design, these fractions naturally sum to 1. plots the partitioning
results for a subset of genes (Figure 1a), and shows a genome-wide
violin plot of the distribution of variance explained by each variable
across all genes (Figure 1b). (Note that these plots show results in
terms of of variance explained, while the results are stored in terms of
the fraction.)
The core functions of variancePartition work seemlessly
with gene expression data stored as a matrix,
data.frame, EList from limma or
ExpressionSet from Biobase.
fitExtractVarPartModel() returns an object that stores the
variance fractions for each gene and each variable in the formula
specified. These fractions can be accessed just like a
data.frame:
# Access first entries
head(varPart)## Batch Individual Tissue Age Residuals
## gene1 0.000157942 0.8903734 0.02468870 4.528911e-05 0.08473466
## gene2 0.000000000 0.8060315 0.10101913 3.336681e-04 0.09261569
## gene3 0.002422410 0.8901149 0.03561726 1.471692e-03 0.07037370
## gene4 0.000000000 0.7688280 0.12531345 1.014413e-03 0.10484417
## gene5 0.000000000 0.6997242 0.20910145 3.871487e-05 0.09113564
## gene6 0.002343666 0.7222283 0.16786542 2.717378e-03 0.10484521
# Access first entries for Individual
head(varPart$Individual)## [1] 0.8903734 0.8060315 0.8901149 0.7688280 0.6997242 0.7222283
# sort genes based on variance explained by Individual
head(varPart[order(varPart$Individual, decreasing = TRUE), ])## Batch Individual Tissue Age Residuals
## gene43 0.000000000 0.9143523 0.011737173 3.776742e-04 0.07353288
## gene174 0.000000000 0.9112702 0.009726000 2.015102e-03 0.07698866
## gene111 0.000000000 0.9067082 0.008386574 9.735963e-04 0.08393164
## gene127 0.000000000 0.9035627 0.013838829 5.081821e-04 0.08209027
## gene151 0.006079206 0.9029393 0.000000000 1.345019e-05 0.09096803
## gene91 0.000000000 0.9002946 0.014144935 1.110517e-06 0.08555939Saving plot to file
In order to save the plot to a file, use the ggsave()
function:
fig <- plotVarPart(vp)
ggsave(file, fig)Plot expression stratified by other variables
variancePartition also includes plotting functions to
visualize the variation across a variable of interest. plots the
expression of a gene stratified by the specified variable. In the
example dataset, users can plot a gene expression trait stratified by
Tissue (Figure 2a) or Individual (Figure 2b).
# get gene with the highest variation across Tissues
# create data.frame with expression of gene i and Tissue
# type for each sample
i <- which.max(varPart$Tissue)
GE <- data.frame(Expression = geneExpr[i, ], Tissue = info$Tissue)
# Figure 2a
# plot expression stratified by Tissue
plotStratify(Expression ~ Tissue, GE, main = rownames(geneExpr)[i])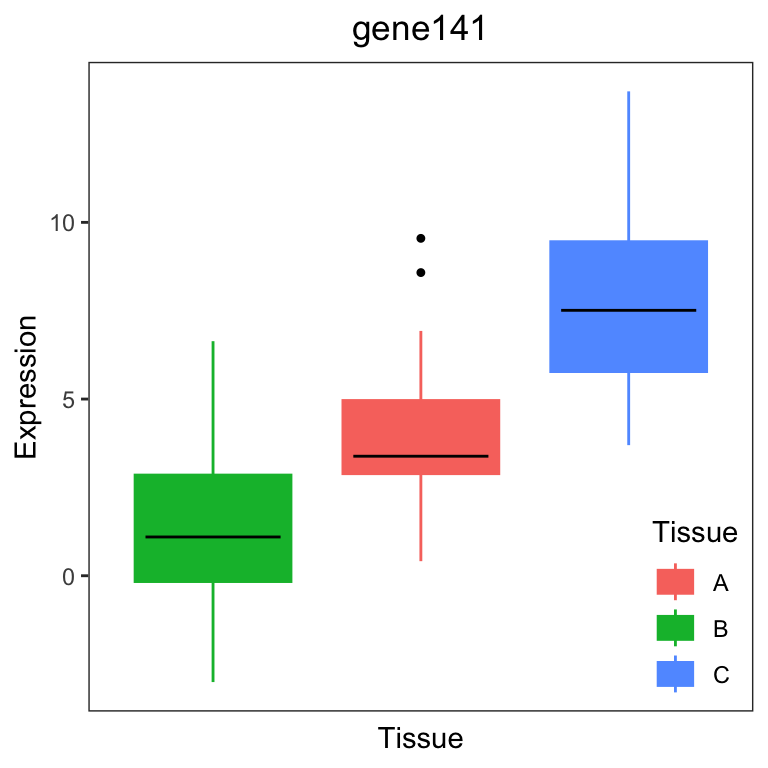
# get gene with the highest variation across Individuals
# create data.frame with expression of gene i and Tissue
# type for each sample
i <- which.max(varPart$Individual)
GE <- data.frame(
Expression = geneExpr[i, ],
Individual = info$Individual
)
# Figure 2b
# plot expression stratified by Tissue
label <- paste("Individual:", format(varPart$Individual[i] * 100,
digits = 3
), "%")
main <- rownames(geneExpr)[i]
plotStratify(Expression ~ Individual, GE,
colorBy = NULL,
text = label, main = main
)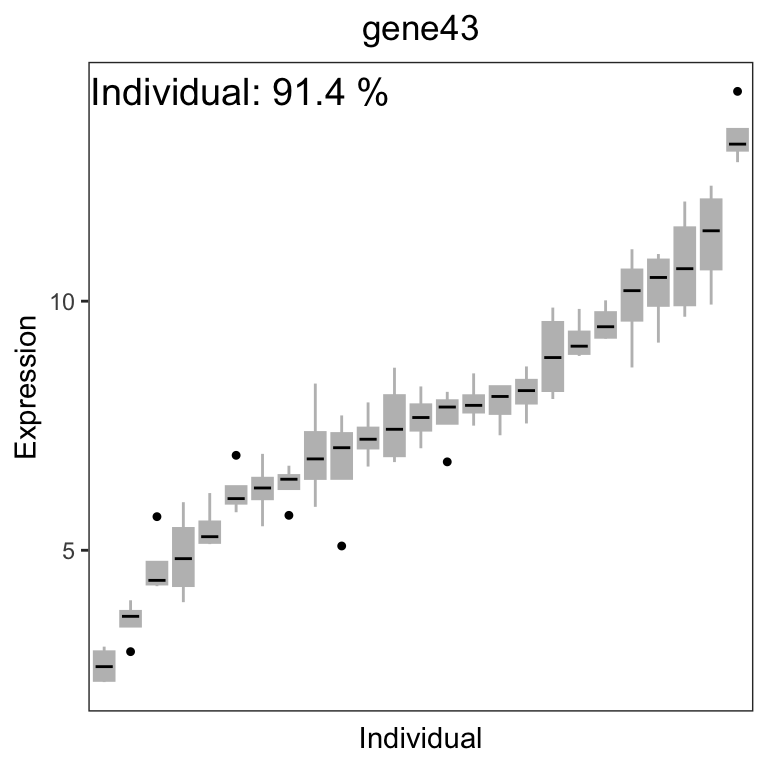
For gene141, variation across tissues explains 52.9% of variance in gene expression. For gene43, variation across Individuals explains 91.4% of variance in gene expression.
Intuition about the backend
At the heart of variancePartition, a regression model is
fit for each gene separately and summary statistics are extracted and
reported to the user for visualization and downstream analysis. For a
single model fit, calcVarPart() computes the fraction of
variance explained by each variable. calcVarPart() is
defined by this package, and computes these statistics from either a
fixed effects model fit with lm() or a linear mixed model
fit with lme4::lmer().
fitExtractVarPartModel() loops over each gene, fits the
regression model and returns the variance fractions reported by
calcVarPart().
Fitting the regression model and extracting variance statistics can also be done directly:
library("lme4")
# fit regression model for the first gene
form_test <- geneExpr[1, ] ~ Age + (1 | Individual) + (1 | Tissue)
fit <- lmer(form_test, info, REML = FALSE)
# extract variance statistics
calcVarPart(fit)## Individual Tissue Age Residuals
## 8.903140e-01 2.468013e-02 4.354738e-05 8.496235e-02Interpretation
variancePartition fits a linear (mixed) model that
jointly considers the contribution of all specified variables on the
expression of each gene. It uses a multiple regression model so that the
effect of each variable is assessed while jointly accounting for all
others. Standard
ANOVA implemented in R involves refitting the model while dropping
terms, but is aimed at hypothesis testing. calcVarPart() is
aimed at estimating variance fractions. It uses a single fit of the
linear (mixed) model and evaluates the sum of squares of each term and
the sum of squares of the total model fit. However, we note that like
any multiple regression model, high correlation bewtween fixed or random
effect variables (see Assess
correlation between all pairs of variables) can produce unstable
estimates and it can be challanging to identify which variable is
responsible for the expression variation.
The results of variancePartition give insight into the
expression data at multiple levels. Moreover, a single statistic often
has multiple equivalent interpretations while only one is relevant to
the biological question. Analysis of the example data in Figure 1 gives
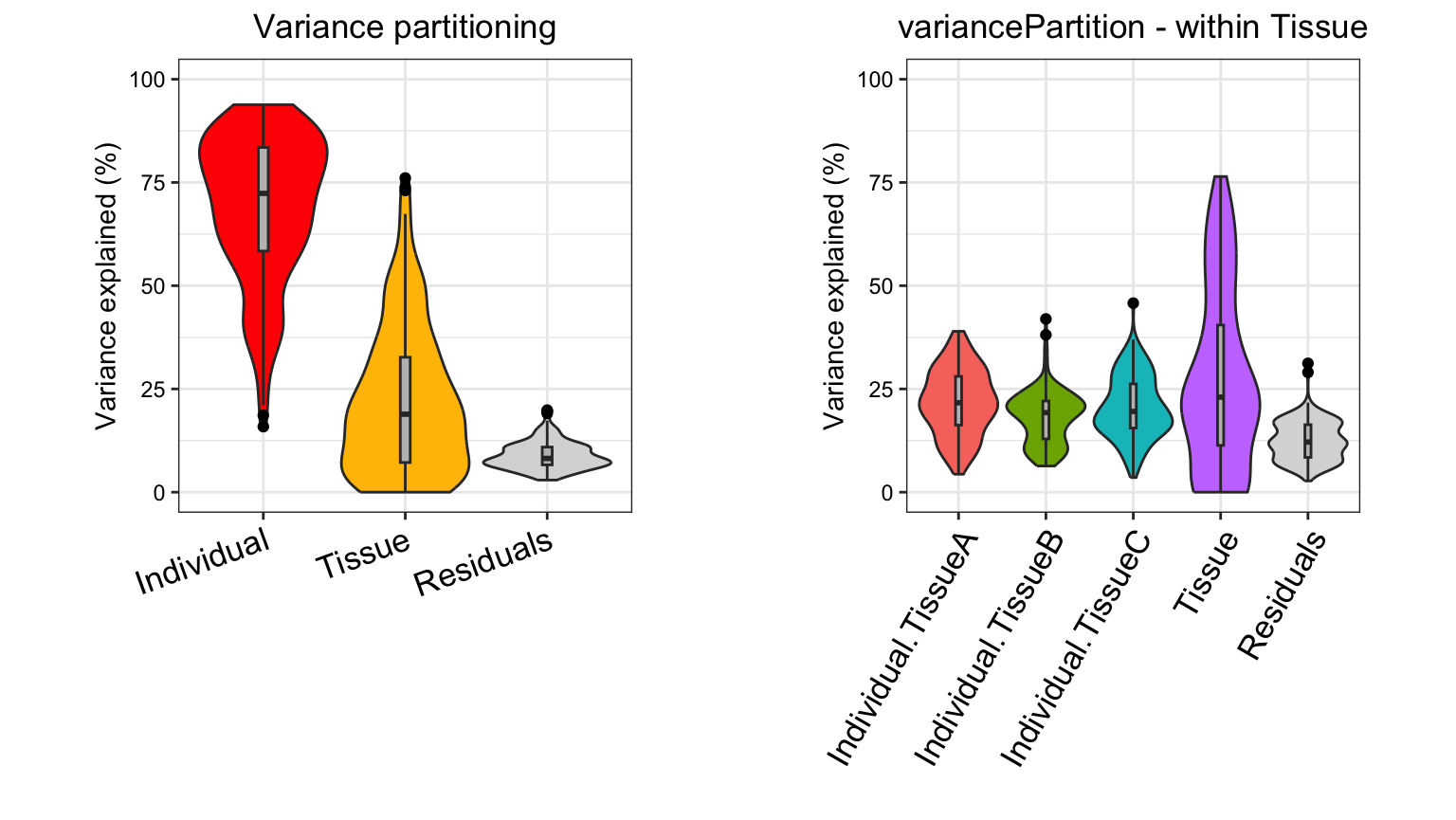
some strong interpretations.
Considering the median across all genes,
- variation across individuals explains a median of 81.5% of the variation in expression, after correcting for tissue, batch and age
- variation across tissues explains a median of 8.2% of the variation in expression, after correcting for other the variables
- variation across batches is negligible after correcting for variation due to other variables
- the effect of age is negligible after correcting for other variables
- correcting for individual, tissue, batch and age leaves a median of 9.3% of the total variance in expression.
These statistics also have a natural interpretation in terms of the intra-class correlation (ICC), the correlation between observations made from samples in the same group.
Considering the median across across all genes and all experiments,
- the ICC for individual is 81.5%.
- the ICC for tissue is 8.2%.
- two randomly selected gene measurements from same individual, but regardless of tissue, batch or age, have a correlation of 81.5%.
- two randomly selected gene measurements from same tissue, but regardless of individual, batch or age, have a correlation of 8.2%.
- two randomly selected gene measurements from the same individual and same tissue, but regardless of batch and age, have an correlation of 81.5% + 8.2% = 89.7%.
Note that that the ICC here is interpreted as the ICC after correcting for all other variables in the model.
These conclusions are based on the genome-wide median across all
genes, but the same type of statements can be made at the gene-level.
Moreover, care must be taken in the interpretation of nested variables.
For example, Age is nested within Individual
since the multiple samples from each individual are taken at the same
age. Thus the effect of Age removes some variation from
being explained by Individual. This often arises when
considering variation across individuals and across sexes: any cross-sex
variation is a component of the cross-individual variation. So the total
variation across individuals is the sum of the fraction of variance
explained by Sex and Individual. This
nesting/summing of effects is common for variables that are properties
of the individual rather than the sample. For example, sex and ethnicity
are always properties of the individual. Variables like age and disease
state can be properties of the individual, but could also vary in
time-course or longitudinal experiments. The the interpretation depends
on the experimental design.
The real power of variancePartition is to identify
specific genes that follow or deviate from the genome-wide trend. The
gene-level statistics can be used to identify a subset of genes that are
enriched for specific biological functions. For example, we can ask if
the 500 genes with the highest variation in expression across tissues
(i.e. the long tail for tissue in Figure 1a) are enriched for genes
known to have high tissue-specificity.
Should a variable be modeled as fixed or random effect?
Categorical variables should (almost) always be modeled as a random
effect. The difference between modeling a categorical variable as a
fixed versus random effect is minimal when the sample size is large
compared to the number of categories (i.e. levels). So variables like
disease status, sex or time point will not be sensitive to modeling as a
fixed versus random effect. However, variables with many categories like
Individual must be modeled as a random effect in
order to obtain statistically valid results. So to be on the safe side,
categorical variable should be modeled as a random effect.
% R and variancePartition handle catagorical variables
stored as a very naturally. If categorical variables are stored as an or
, they must be converted to a before being used with
variancePartition
variancePartition fits two types of models:
linear mixed model where all categorical variables are modeled as random effects and all continuous variables are fixed effects. The function
lme4::lmer()is used to fit this model.fixed effected model, where all variables are modeled as fixed effects. The function
lm()is used to fit this model.
Which variables should be included?
In my experience, it is useful to include all variables in the first
analysis and then drop variables that have minimal effect. However, like
all multiple regression methods, variancePartition will
divide the contribution over multiple variables that are strongly
correlated. So, for example, including both sex and height in the model
will show sex having a smaller contribution to variation gene expression
than if height were omitted, since there variables are strongly
correlated. This is a simple example, but should give some intuition
about a common issue that arises in analyses with
variancePartition.
variancePartition can naturally assess the contribution
of both individual and sex in a dataset. As expected, genes for which
sex explains a large fraction of variation are located on chrX and chrY.
If the goal is to interpret the impact of sex, then there is no issue.
But recall the issue with correlated variables and note that individual
is correlated with sex, because each individual is only one sex
regardless of how many samples are taken from a individual. It follows
that including sex in the model reduces the apparent contribution
of individual to gene expression. In other words, the ICC for individual
will be different if sex is included in the model.
In general, including variables in the model that do not vary within
individual will reduce the apparent contribution of individual as
estimated by variancePartition. For example, sex and
ethnicity never vary between multiple samples from the same individual
and will always reduce the apparent contribution of individual. However,
disease state and age may or may not vary depending on the study
design.
In biological datasets technical variability (i.e. batch effects) can
often reduce the apparent biological signal. In RNA-seq analysis, it is
common for the the impact of this technical variability to be removed
before downstream analysis. Instead of including these batch variable in
the variancePartition analysis, it is simple to complete
the expression residuals with the batch effects removed and then feeds
these residuals to variancePartition. This will increase
the fraction of variation explained by biological variables since
technical variability is reduced.
Assess correlation between all pairs of variables
Evaluating the correlation between variables in a important part in interpreting variancePartition results. When comparing two continuous variables, Pearson correlation is widely used. But variancePartition includes categorical variables in the model as well. In order to accommodate the correlation between a continuous and a categorical variable, or two categorical variables we used canonical correlation analysis.
Canonical Correlation Analysis (CCA) is similar to correlation
between two vectors, except that CCA can accommodate matricies as well.
For a pair of variables, canCorPairs() assesses the degree
to which they co-vary and contain the same information. Variables in the
formula can be a continuous variable or a discrete variable expanded to
a matrix (which is done in the backend of a regression model). For a
pair of variables, canCorPairs() uses CCA to compute the
correlation between these variables and returns the pairwise correlation
matrix.
Statistically, let rho be the array of correlation
values returned by the standard R function cancor to compute CCA.
canCorPairs() returns rho / sum(rho) which is
the fraction of the maximum possible correlation. Note that CCA returns
correlations values between 0 and 1
form <- ~ Individual + Tissue + Batch + Age + Height
# Compute Canonical Correlation Analysis (CCA)
# between all pairs of variables
# returns absolute correlation value
C <- canCorPairs(form, info)## Warning: the 'subbars' function has moved to the reformulas package. Please update your imports, or ask an upstream package maintainter to do so.
## This warning is displayed once per session.
# Plot correlation matrix
# between all pairs of variables
plotCorrMatrix(C)
Advanced analysis
Extracting additional information from model fits
Advanced users may want to perform the model fit and extract results in separate steps in order to examine the fit of the model for each gene. Thus the work of can be divided into two steps: 1) fit the regression model, and 2) extracting variance statistics.
form <- ~ Age + (1 | Individual) + (1 | Tissue) + (1 | Batch)
# Fit model
results <- fitVarPartModel(geneExpr, form, info)
# Extract results
varPart <- extractVarPart(results)Note that storing the model fits can use a lot of memory (~10Gb with 20K genes and 1000 experiments). I do not recommend unless you have a specific need for storing the entire model fit.
Instead, fitVarPartModel() can extract any desired
information using any function that accepts the model fit from
lm() or lmer(). The results are stored in a
list and can be used for downstream analysis.
# Fit model and run summary() function on each model fit
vpSummaries <- fitVarPartModel(geneExpr, form, info, fxn = summary)
# Show results of summary() for the first gene
vpSummaries[[1]]## Linear mixed model fit by maximum likelihood ['lmerMod']
## Formula: y.local ~ Age + (1 | Individual) + (1 | Tissue) + (1 | Batch)
## Data: data
## Weights: data$w.local
## Control: control
##
## AIC BIC logLik -2*log(L) df.resid
## 397.2 412.8 -192.6 385.2 94
##
## Scaled residuals:
## Min 1Q Median 3Q Max
## -2.05797 -0.58651 0.01466 0.66030 1.97077
##
## Random effects:
## Groups Name Variance Std.Dev.
## Individual (Intercept) 10.82274 3.28979
## Batch (Intercept) 0.00192 0.04382
## Tissue (Intercept) 0.30010 0.54781
## Residual 1.02997 1.01488
## Number of obs: 100, groups: Individual, 25; Batch, 4; Tissue, 3
##
## Fixed effects:
## Estimate Std. Error t value
## (Intercept) -10.602426 1.094925 -9.683
## Age 0.003183 0.016103 0.198
##
## Correlation of Fixed Effects:
## (Intr)
## Age -0.739Removing batch effects before fitting model
Gene expression studies often have substantial batch effects, and
variancePartition can be used to understand the magnitude
of the effects. However, we often want to focus on biological variables
(i.e. individual, tissue, disease, sex) after removing the effect of
technical variables. Depending on the size of the batch effect, I have
found it useful to correct for the batch effect first and then perform a
variancePartition analysis afterward. Subtracting this
batch effect can reduce the total variation in the data, so that the
contribution of other variables become clearer.
Standard analysis:
form <- ~ (1 | Tissue) + (1 | Individual) + (1 | Batch) + Age
varPart <- fitExtractVarPartModel(geneExpr, form, info)Analysis on residuals:
library("limma")
# subtract out effect of Batch
fit <- lmFit(geneExpr, model.matrix(~Batch, info))
res <- residuals(fit, geneExpr)
# fit model on residuals
form <- ~ (1 | Tissue) + (1 | Individual) + Age
varPartResid <- fitExtractVarPartModel(res, form, info)Remove batch effect with linear mixed model
# subtract out effect of Batch with linear mixed model
modelFit <- fitVarPartModel(geneExpr, ~ (1 | Batch), info)
res <- residuals(modelFit)
# fit model on residuals
form <- ~ (1 | Tissue) + (1 | Individual) + Age
varPartResid <- fitExtractVarPartModel(res, form, info)If the two-step process requires too much memory, the residuals can
be computed more efficiently. Here, run the function inside the call to
fitVarPartModel() to avoid storing the large intermediate
results.
# extract residuals directly without storing intermediate results
residList <- fitVarPartModel(geneExpr, ~ (1 | Batch), info,
fxn = residuals
)
# convert list to matrix
residMatrix <- do.call(rbind, residList)Variation within multiple subsets of the data
So far, we have focused on interpreting one variable at a time. But
the linear mixed model behind variancePartition is a very
powerful framework for analyzing variation at multiple levels. We can
easily extend the previous analysis of the contribution of individual
and tissue on variation in gene expression to examine the contribution
of individual within each tissue. This analysis is as easy as
specifying a new formula and rerunning variancePartition.
Note that is analysis will only work when there are replicates for at
least some individuals within each tissue in order to assess
cross-individual variance with in a tissue.
# specify formula to model within/between individual variance
# separately for each tissue
# Note that including +0 ensures each tissue is modeled explicitly
# Otherwise, the first tissue would be used as baseline
form <- ~ (Tissue + 0 | Individual) + Age + (1 | Tissue) + (1 | Batch)
# fit model and extract variance percents
varPart <- fitExtractVarPartModel(geneExpr, form, info, showWarnings = FALSE)
# violin plot
plotVarPart(sortCols(varPart), label.angle = 60)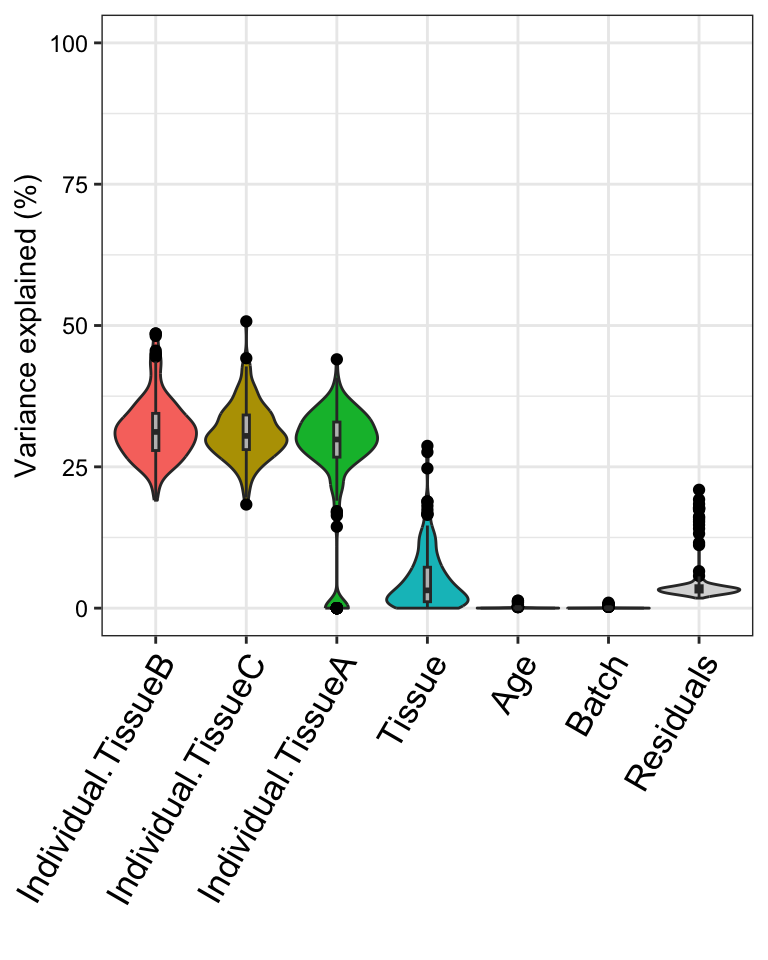
This analysis corresponds to a varying coefficient model, where the
correlation between individuals varies for each tissue ]. Since the
variation across individuals is modeled within each tissue, the total
variation explained does not sum to 1 as it does for standard
application of variancePartition. So interpretation as
intra-class does not strictly apply and use of
plotPercentBars() is no longer applicable. Yet the
variables in the study design are still ranked in terms of their
genome-wide contribution to expression variation, and results can still
be analyzed at the gene level. See Variation with
multiple subsets of the data for statistical details.
Detecting problems caused by collinearity of variables
Including variables that are highly correlated can produce misleading
results and overestimate the contribution of variables modeled as fixed
effects. This is usually not an issue, but can arise when statistically
redundant variables are included in the model. In this case, the model
is "degenerate" or "computationally singular"
and parameter estimates from this model are not meaningful. Dropping one
or more of the covariates will fix this problem.
A check of collinearity is built into fitVarPartModel()
and fitExtractVarPartModel(), so the user will be warned if
this is an issue.
Alternatively, the user can use the colinearityScore()
function to evaluate whether this is an issue for a single model
fit:
form <- ~ (1 | Individual) + (1 | Tissue) + Age + Height
# fit model
res <- fitVarPartModel(geneExpr[1:4, ], form, info)
# evaluate the collinearity score on the first model fit
# this reports the correlation matrix between coefficient estimates
# for fixed effects
# the collinearity score is the maximum absolute correlation value
# If the collinearity score > .99 then the variance partition
# estimates may be problematic
# In that case, a least one variable should be omitted
colinearityScore(res[[1]])## [1] 0.7774082
## attr(,"vcor")
## (Intercept) Age Height
## (Intercept) 1.0000000 -0.41907978 -0.77740819
## Age -0.4190798 1.00000000 -0.05748224
## Height -0.7774082 -0.05748224 1.00000000Including weights computed separately
variancePartition automatically used precision weights
computed by limma::voom(), but the user can also specify
custom weights using the weightsMatrix argument.
form <- ~ (1 | Individual) + (1 | Tissue) + Age + Height
# Specify custom weights
# In this example the weights are simulated from a
# uniform distribution and are not meaningful.
weights <- matrix(runif(length(geneExpr)), nrow = nrow(geneExpr))
# Specify custom weights
res <- fitExtractVarPartModel(geneExpr[1:4, ], form, info,
weightsMatrix = weights[1:4, ]
)In addition, setting the useWeights=FALSE will suppress
usage of the weights in all cases, i.e. when the weights are specified
manually or implicitly with the results of
limma::voom().
Including interaction terms
Typical analysis assumes that the effect of each variable on gene
expression does not depend on other variables in the model. Sometimes
this assumption is too strict, and we want to model an interaction
effect whereby the effect of Batch depends on
Tissue. This can be done easly by specifying an interaction
term, (1|Batch:Tissue). Since Batch has 4
categories and Tissue has 3, this interaction term
implicity models a new 3*4 = 12 category variable in the
analysis. This new interaction term will absorb some of the variance
from the Batch and Tissue term, so an
interaction model should always include the two constituent
variables.
Here we fit an interaction model, but we observe that interaction
between Batch and Tissue does not explain much
expression variation.
form <- ~ (1 | Individual) + Age + Height + (1 | Tissue) + (1 | Batch) +
(1 | Batch:Tissue)
# fit model
vpInteraction <- fitExtractVarPartModel(geneExpr, form, info)
plotVarPart(sortCols(vpInteraction))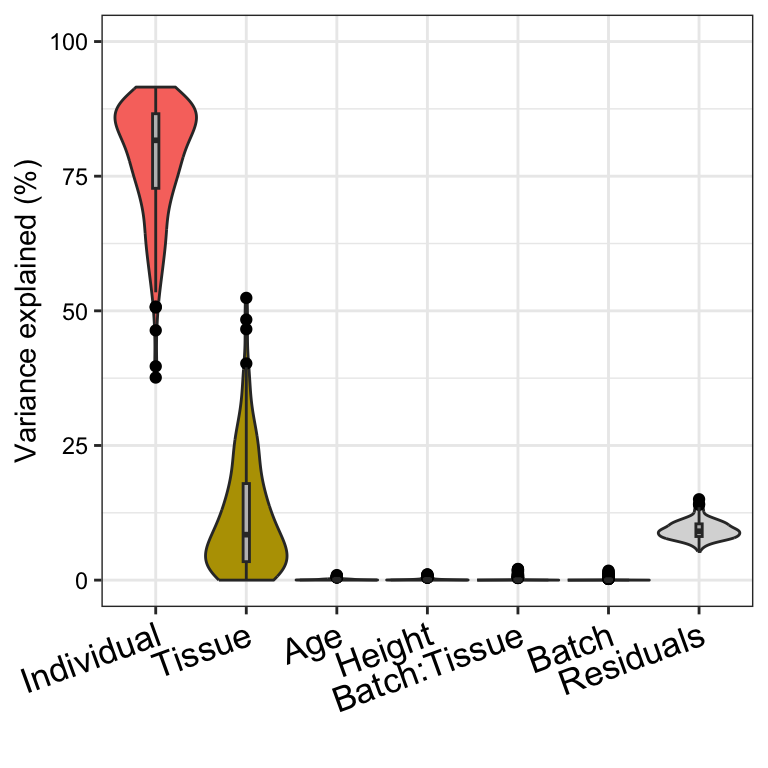
Application to expression data
variancePartition works with gene expression data that
has already been processed and normalized as for differential expression
analysis.
Gene-level counts
featureCounts (Liao et al.
2014) and HTSeq (Anders et
al. 2015) report the number of reads mapping to each gene (or
exon). These results are easily read into R. limma::voom()
and DESeq2 are widely used for differential expression
analysis of gene- and exon-level counts and can be used to process data
before analysis with variancePartition. This section
addresses processing and normalization of gene-level counts, but the
analysis is the same for exon-level counts.
limma::voom()
Read RNA-seq counts into R, normalize for library size within and
between experiments with TMM (Robinson and
Oshlack 2010), estimate precision weights with
limma::voom().
library("limma")
library("edgeR")
# identify genes that pass expression cutoff
isexpr <- rowSums(cpm(geneCounts) > 1) >= 0.5 * ncol(geneCounts)
# create data structure with only expressed genes
gExpr <- DGEList(counts = geneCounts[isexpr, ])
# Perform TMM normalization
gExpr <- calcNormFactors(gExpr)
# Specify variables to be included in the voom() estimates of
# uncertainty.
# Recommend including variables with a small number of categories
# that explain a substantial amount of variation
design <- model.matrix(~Batch, info)
# Estimate precision weights for each gene and sample
# This models uncertainty in expression measurements
vobjGenes <- voom(gExpr, design)
# Define formula
form <- ~ (1 | Individual) + (1 | Tissue) + (1 | Batch) + Age
# variancePartition seamlessly deals with the result of voom()
# by default, it seamlessly models the precision weights
# This can be turned off with useWeights=FALSE
varPart <- fitExtractVarPartModel(vobjGenes, form, info)
DESeq2
Process and normalize the gene-level counts before running
variancePartition analysis.
library("DESeq2")
# create DESeq2 object from gene-level counts and metadata
dds <- DESeqDataSetFromMatrix(
countData = geneCounts,
colData = info,
design = ~1
)## Warning in S4Vectors:::anyMissing(runValue(x_seqnames)): 'S4Vectors:::anyMissing()' is deprecated.
## Use 'anyNA()' instead.
## See help("Deprecated")## Warning in S4Vectors:::anyMissing(runValue(strand(x))): 'S4Vectors:::anyMissing()' is deprecated.
## Use 'anyNA()' instead.
## See help("Deprecated")
# Estimate library size correction scaling factors
dds <- estimateSizeFactors(dds)
# identify genes that pass expression cutoff
isexpr <- rowSums(fpm(dds) > 1) >= 0.5 * ncol(dds)
# compute log2 Fragments Per Million
# Alternatively, fpkm(), vst() or rlog() could be used
quantLog <- log2(fpm(dds)[isexpr, ] + 1)
# Define formula
form <- ~ (1 | Individual) + (1 | Tissue) + (1 | Batch) + Age
# Run variancePartition analysis
varPart <- fitExtractVarPartModel(quantLog, form, info)Note that DESeq2 does not compute precision weights like
limma::voom(), so they are not used in this version of the
analysis.
Isoform quantification
Other software performs isoform-level quantification using reads that
map to multiple transcripts. These include kallisto (Bray et al. 2016), sailfish (Patro et al. 2014), salmon Patro et al. (2015), RSEM (Li and Dewey 2011)and cufflinks
(Trapnell et al. 2010.)
tximport
Quantifications from kallisto, salmon,
sailfish and RSEM can be read into R and
processed with the Bioconductor package tximport. The gene-
or transcript-level quantifications can be used directly in
variancePartition.
library("tximportData")
library("tximport")
library("readr")
# Get data from folder where tximportData is installed
dir <- system.file("extdata", package = "tximportData")
samples <- read.table(file.path(dir, "samples.txt"), header = TRUE)
files <- file.path(dir, "kallisto", samples$run, "abundance.tsv")
names(files) <- paste0("sample", 1:6)
tx2gene <- read.csv(file.path(dir, "tx2gene.csv"))
# reads results from kallisto
txi <- tximport(files,
type = "kallisto", tx2gene = tx2gene,
countsFromAbundance = "lengthScaledTPM"
)
# define metadata (usually read from external source)
info_tximport <- data.frame(
Sample = sprintf("sample%d", 1:6),
Disease = c("case", "control")[c(rep(1, 3), rep(2, 3))]
)
# Extract counts from kallisto
y <- DGEList(txi$counts)
# compute library size normalization
y <- calcNormFactors(y)
# apply voom to estimate precision weights
design <- model.matrix(~Disease, data = info_tximport)
vobj <- voom(y, design)
# define formula
form <- ~ (1 | Disease)
# Run variancePartition analysis (on only 10 genes)
varPart_tx <- fitExtractVarPartModel(
vobj[1:10, ], form,
info_tximport
)Code to process results from sailfish,
salmon, RSEM is very similar.
See tutorial for more details.
ballgown
Quantifications from Cufflinks/Tablemaker and RSEM can be processed
and read into R with the Bioconductor package ballgown.
library("ballgown")
# Get data from folder where ballgown is installed
data_directory <- system.file("extdata", package = "ballgown")
# Load results of Cufflinks/Tablemaker
bg <- ballgown(
dataDir = data_directory, samplePattern = "sample",
meas = "all"
)## Warning in S4Vectors:::anyMissing(runValue(x_seqnames)): 'S4Vectors:::anyMissing()' is deprecated.
## Use 'anyNA()' instead.
## See help("Deprecated")## Warning in S4Vectors:::anyMissing(runValue(strand(x))): 'S4Vectors:::anyMissing()' is deprecated.
## Use 'anyNA()' instead.
## See help("Deprecated")## Warning in S4Vectors:::anyMissing(runValue(x_seqnames)): 'S4Vectors:::anyMissing()' is deprecated.
## Use 'anyNA()' instead.
## See help("Deprecated")## Warning in S4Vectors:::anyMissing(runValue(strand(x))): 'S4Vectors:::anyMissing()' is deprecated.
## Use 'anyNA()' instead.
## See help("Deprecated")
# extract gene-level FPKM quantification
# Expression can be convert to log2-scale if desired
gene_expression <- gexpr(bg)
# extract transcript-level FPKM quantification
# Expression can be convert to log2-scale if desired
transcript_fpkm <- texpr(bg, "FPKM")
# define metadata (usually read from external source)
info_ballgown <- data.frame(
Sample = sprintf("sample%02d", 1:20),
Batch = rep(letters[1:4], 5),
Disease = c("case", "control")[c(rep(1, 10), rep(2, 10))]
)
# define formula
form <- ~ (1 | Batch) + (1 | Disease)
# Run variancePartition analysis
# Gene-level analysis
varPart_gene <- fitExtractVarPartModel(
gene_expression, form,
info_ballgown
)## Warning in filterInputData(exprObj, formula, data, useWeights = useWeights): Sample names of responses (i.e. columns of exprObj) do not match
## sample names of metadata (i.e. rows of data). Recommend consistent
## names so downstream results are labeled consistently.## Warning in .fitExtractVarPartModel(exprObj, formula, data, REML = REML, : Model failed for 1 responses.
## See errors with attr(., 'errors')
# Transcript-level analysis
varPart_transcript <- fitExtractVarPartModel(
transcript_fpkm, form,
info_ballgown
)## Warning in filterInputData(exprObj, formula, data, useWeights = useWeights): Sample names of responses (i.e. columns of exprObj) do not match
## sample names of metadata (i.e. rows of data). Recommend consistent
## names so downstream results are labeled consistently.## Warning in .fitExtractVarPartModel(exprObj, formula, data, REML = REML, : Model failed for 2 responses.
## See errors with attr(., 'errors')Note that ballgownrsem can be used for a similar
analysis of RSEM results.
See tutorial for more details.
Compare with other methods
Characterizing drivers of variation in gene expression data has
typically relied on principal components analysis (PCA) and hierarchical
clustering. Here I apply these methods to two simulated datasets to
demonstrate the additional insight from an analysis with
variancePartition. Each simulated dataset comprises 60
experiments from 10 individuals and 3 tissues with 2 biological
replicates. In the first dataset, tissue is the major driver of
variation in gene expression(Figure @ref(fig:siteDominant)). In the
second dataset, individual is the major driver of variation in gene
expression (Figures @ref(fig:IndivDominant)).
Analysis of simulated data illustrates that PCA identifies the major driver of variation when tissue is dominant and there are only 3 categories. But the results are less clear when individual is dominant because there are now 10 categories. Meanwhile, hierarchical clustering identifies the major driver of variation in both cases, but does not give insight into the second leading contributor.
Analysis with variancePartition has a number of
advantages over these standard methods:
-
variancePartitionprovides a natural interpretation of multiple variables- figures from PCA/hierarchical clustering allow easy interpretation of only one variable
-
variancePartitionquantifies the contribution of each variable- PCA/hierarchical clustering give only a visual representation
-
variancePartitioninterprets contribution of each variable to each gene individually for downstream analysis- PCA/hierarchical clustering produces genome-wide summary and does not allow gene-level interpretation
variancePartitioncan assess contribution of one variable (i.e. Individual) separately in subset of the data defined by another variable (i.e. Tissue)
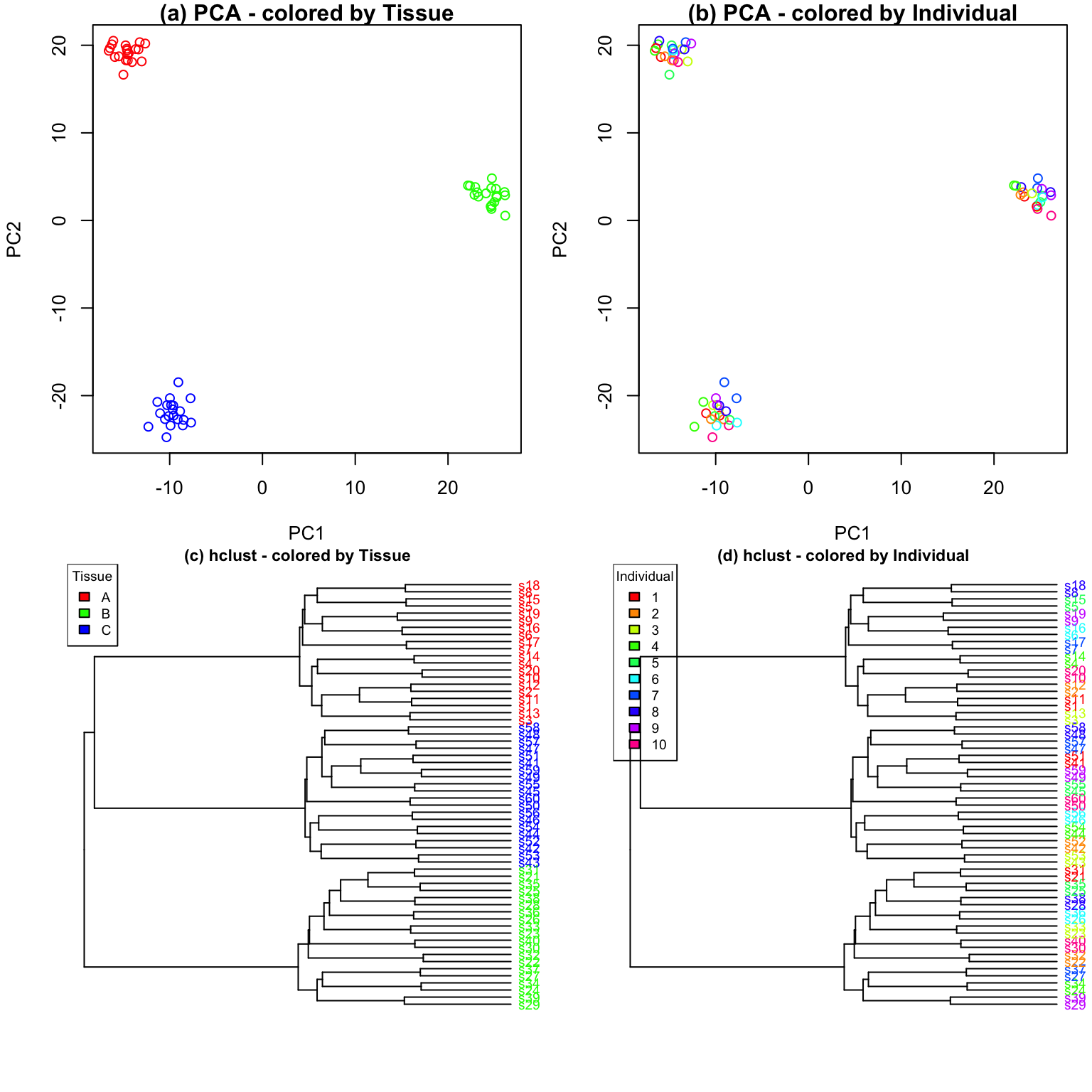
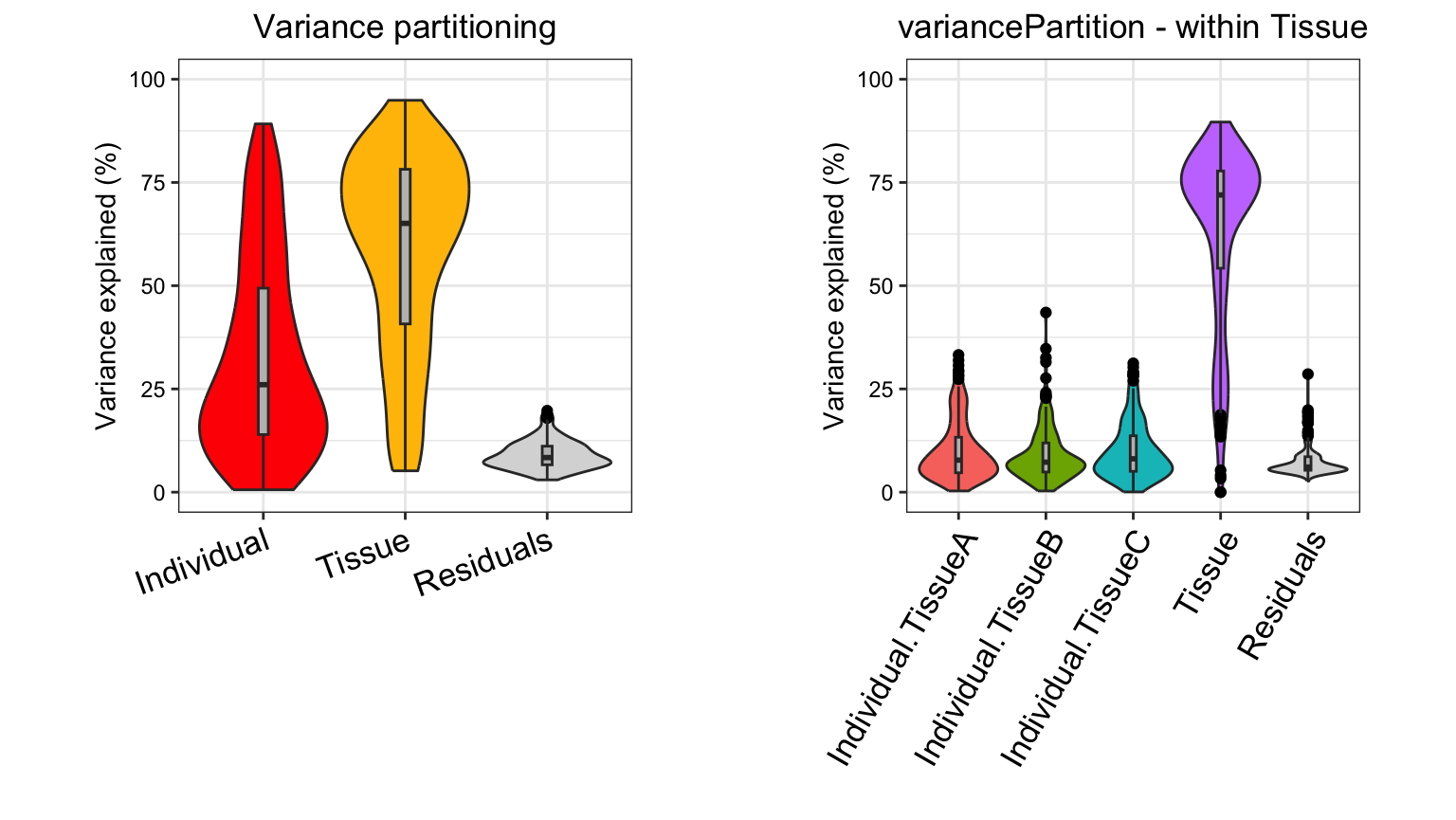
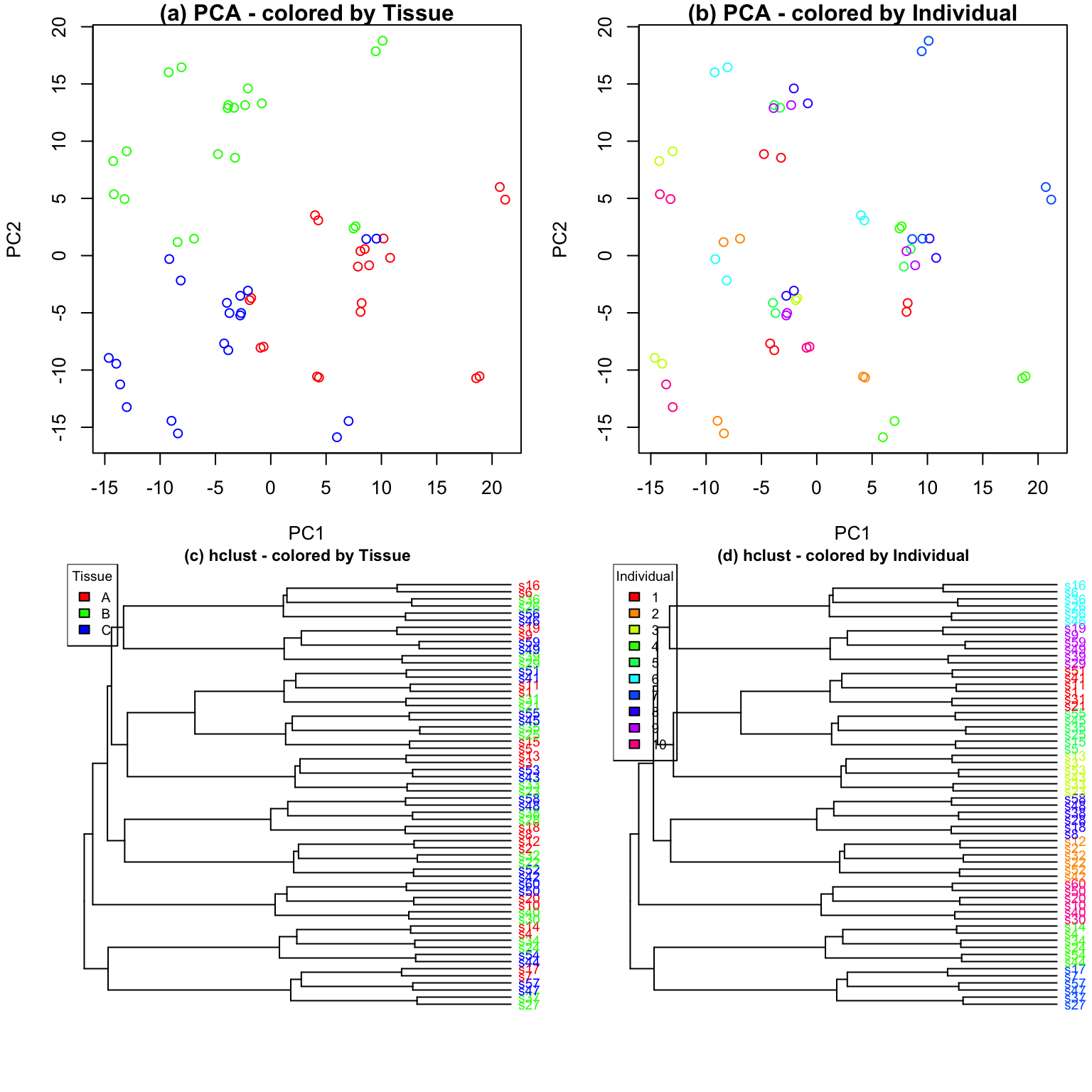
Statistical details
A variancePartition analysis evaluates the linear
(mixed) model \begin{eqnarray}
y &=& \sum_j X_j\beta_j + \sum_k Z_k \alpha_k + \varepsilon \\
\alpha_k &\sim& \mathcal{N}(0, \sigma^2_{\alpha_k})\\
\varepsilon &\sim& \mathcal{N}(0, \sigma^2_\varepsilon)
\end{eqnarray} where y is the
expression of a single gene across all samples, X_j is the matrix of j^{th} fixed effect with coefficients \beta_j, Z_k
is the matrix corresponding to the k^{th} random effect with coefficients \alpha_k drawn from a normal distribution
with variance \sigma^2_{\alpha_k}. The
noise term, \varepsilon, is drawn from
a normal distribution with variance \sigma^2_\varepsilon. Parameters are
estimated with maximum likelihood, rather than REML, so that fixed
effect coefficients, \beta_j, are
explicitly estimated.
I use the term “linear (mixed) model” here since
variancePartition works seamlessly when a fixed effects
model (i.e. linear model) is specified.
Variance terms for the fixed effects are computed using the post
hoc calculation
\begin{eqnarray}
\hat{\sigma}^2_{\beta_j} = \text{var}\left( X_j \hat{\beta}_j\right).
\end{eqnarray} For a fixed effects model, this corresponds to the
sum of squares for each component of the model.
For a standard application of the linear mixed model, where the effect of each variable is additive, the fraction of variance explained by the j^{th} fixed effect is \begin{eqnarray} \frac{\hat{\sigma}^2_{\beta_j}}{\sum_j \hat{\sigma}^2_{\beta_j} + \sum_k \hat{\sigma}^2_{\alpha_k} + \hat{\sigma}^2_\varepsilon}, \end{eqnarray} by the k^{th} random effect is \begin{eqnarray} \frac{\hat{\sigma}^2_{\alpha_k}}{\sum_j \hat{\sigma}^2_{\beta_j} + \sum_k \hat{\sigma}^2_{\alpha_k} + \hat{\sigma}^2_\varepsilon}, \end{eqnarray} and the residual variance is \begin{eqnarray} \frac{\hat{\sigma}^2_{\varepsilon}}{\sum_j \hat{\sigma}^2_{\beta_j} + \sum_k \hat{\sigma}^2_{\alpha_k} + \hat{\sigma}^2_\varepsilon}. \end{eqnarray}
Implementation in R
An R formula is used to define the terms in the fixed and random
effects, and fitVarPartModel() fits the specified model for
each gene separately. If random effects are specified,
lme4::lmer() is used behind the scenes to fit the model,
while lm() is used if there are only fixed effects.
fitVarPartModel() returns a list of the model fits, and
returns the variance partition statistics for each model in the list.
fitExtractVarPartModel() combines the actions of
fitVarPartModel() and into one function call.
calcVarPart() is called behind the scenes to compute
variance fractions for both fixed and mixed effects models, but the user
can also call this function directly on a model fit with
lm() or lmer().
Interpretation of percent variance explained
The percent variance explained can be interpreted as the intra-class correlation (ICC) when a special case of Equation 1 is used. Consider the simplest example of the i^{th} sample from the k^{th} individual \begin{eqnarray} y_{i,k} = \mu + Z \alpha_{i,k} + e_{i,k} \end{eqnarray} with only an intercept term, one random effect corresponding to individual, and an error term. In this case ICC corresponds to the correlation between two samples from the same individual. This value is equal to the fraction of variance explained by individual. For example, consider the correlation between samples from the same individual: \begin{eqnarray} ICC &=& cor( y_{1,k}, y_{2,k}) \\ &=& cor( \mu + Z \alpha_{1,k} + e_{1,k}, \mu + Z \alpha_{2,k} + e_{2,k}) \\ &=& \frac{cov( \mu + Z \alpha_{1,k} + e_{1,k}, \mu + Z \alpha_{2,k} + e_{2,k})}{ \sqrt{ var(\mu + Z \alpha_{1,k} + e_{1,k}) var( \mu + Z \alpha_{2,k} + e_{2,k})}}\\ &=& \frac{cov(Z \alpha_{1,k}, Z \alpha_{2,k})}{\sigma^2_\alpha + \sigma^2_\varepsilon} \\ &=& \frac{\sigma^2_\alpha}{\sigma^2_\alpha + \sigma^2_\varepsilon} \end{eqnarray} The correlation between samples from different individuals is: \begin{eqnarray} &=& cor( y_{1,1}, y_{1,2}) \\ &=& cor( \mu + Z \alpha_{1,1} + e_{1,1}, \mu + Z \alpha_{1,2} + e_{1,2}) \\ &=& \frac{cov(Z \alpha_{1,1}, Z \alpha_{1,2})}{\sigma^2_\alpha + \sigma^2_\varepsilon} \\ &=& \frac{0}{\sigma^2_\alpha + \sigma^2_\varepsilon} \\ &=& 0 \end{eqnarray} This interpretation in terms of fraction of variation explained (FVE) naturally generalizes to multiple variance components. Consider two sources of variation, individual and cell type with variances \sigma^2_{id} and \sigma^2_{cell}, respectively. Applying a generalization of the the previous derivation, two samples are correlated according to:
| Individual | cell type | variance | Interpretation | Correlation value |
|---|---|---|---|---|
| same | different | \frac{\sigma^2_{id}}{\sigma^2_{id} + \sigma^2_{cell} + \sigma^2_\varepsilon } | FVE by individual | ICC_{individual} |
| different | same | \frac{\sigma^2_{cell}}{\sigma^2_{id} + \sigma^2_{cell} + \sigma^2_\varepsilon } | FVE by cell type | ICC_{cell} |
| same | same | \frac{\sigma^2_{id} + \sigma^2_{cell}}{\sigma^2_{id} + \sigma^2_{cell} + \sigma^2_\varepsilon } | sum of FVE by individual & cell type | ICC_{individual,cell} |
| different | different | \frac{0}{\sigma^2_{id} + \sigma^2_{cell} + \sigma^2_\varepsilon } | sample are independent |
Notice that the correlation between samples from the same individual and same cell type corresponds to the sum of the fraction explained by individual + fraction explained by cell type. This defines ICC for individual and tissue, as well as the combined ICC and relates these values to FVE.
In order to illustrate how this FVE and ICC relate to the correlation between samples in multilevel datasets, consider a simple example of 5 samples from 2 individuals and 2 tissues:
| Sample | Individual | Cell type |
|---|---|---|
| a | 1 | T-Cell |
| b | 1 | T-Cell |
| c | 1 | monocyte |
| d | 2 | T-Cell |
| e | 2 | monocyte |
Modeling the separate effects of individual and tissue gives the following covariance structure between samples when a linear mixed model is used:
\begin{array}{ccc} & \\ cov(y)=\begin{array}{ccccc} a \\ b \\ c\\ d\\ e\end{array} & \left( \begin{array}{ccccc} \sigma^2_{id} + \sigma^2_{cell} + \sigma^2_\varepsilon & & &&\\ \sigma^2_{id} + \sigma^2_{cell} & \sigma^2_{id} + \sigma^2_{cell} + \sigma^2_\varepsilon & & &\\ \sigma^2_{id} & \sigma^2_{id} &\sigma^2_{id} + \sigma^2_{cell} + \sigma^2_\varepsilon & &\\ \sigma^2_{cell} & \sigma^2_{cell} &0 &\sigma^2_{id} + \sigma^2_{cell} + \sigma^2_\varepsilon &\\ 0 & 0 &\sigma^2_{cell} & \sigma^2_{id} &\sigma^2_{id} + \sigma^2_{cell} + \sigma^2_\varepsilon \\ \end{array} \right)\end{array}
The covariance matrix is symmetric so that blank entries take the value on the opposite side of the diagonal. The covariance can be converted to correlation by dividing by \sigma^2_{id} + \sigma^2_{cell} + \sigma^2_\varepsilon, and this gives the results from above. This example generalizes to any number of variance components (Pinheiro and Bates 2000).
Variation with multiple subsets of the data
The linear mixed model underlying variancePartition
allows the effect of one variable to depend on the value of another
variable. Statistically, this is called a varying coefficient model
(Pinheiro and Bates 2000; Galecki and Burzykowski
2013). This model arises in variancePartition
analysis when the variation explained by individual depends on tissue or
cell type.
A given sample is only from one cell type, so this analysis asks a question about a subset of the data. The the data is implicitly divided into subsets base on cell type and variation explained by individual is evaluated within each subset. The data is not actually divided onto subset, but the statistical model essentially examples samples with each cell type. This subsetting means that the variance fractions do not sum to 1.
Consider a concrete example with variation from across individual and cell types (T-cells and monocytes) with data from the i^{th} sample from the k^{th} individual, sex of s and cell type c. Modeling the variation across individuals within cell type corresponds to \begin{eqnarray} y_{i,k,s,c} = \mu + Z^{(sex)} \alpha_{i,s} + Z^{(Tcell| id)} \alpha_{i,k,c} + Z^{(monocyte| id)} \alpha_{i,k,c} + e_{i,k,s,c} \end{eqnarray}
with corresponding variance components:
| Variance component | Interpretation |
|---|---|
| \sigma^2_{sex} | variance across sex (which is the same for all cell types) |
| \sigma^2_{(Tcell| id)} | variance across individuals within T-cells |
| \sigma^2_{(monocyte| id)} | variance across individuals within monocytes |
| \sigma^2_\varepsilon | residual variance |
Since the dataset is now divided into multiple subsets, direct
interpretation of the fraction of variation explained (FVE) as
intra-class correlation does not apply. Instead, we compute a
“pseudo-FVE” by approximating the total variance attributable to cell
type by using a weighted average of the within cell type variances
weighted by the sample size within each cell type. Thus the values of
pseudo-FVE do not have the simple interpretation as in the standard
application of variancePartition, but allows ranking of
variables based on genome-wide contribution to variance and analysis of
gene-level results.
Variance partitioning and differential expression
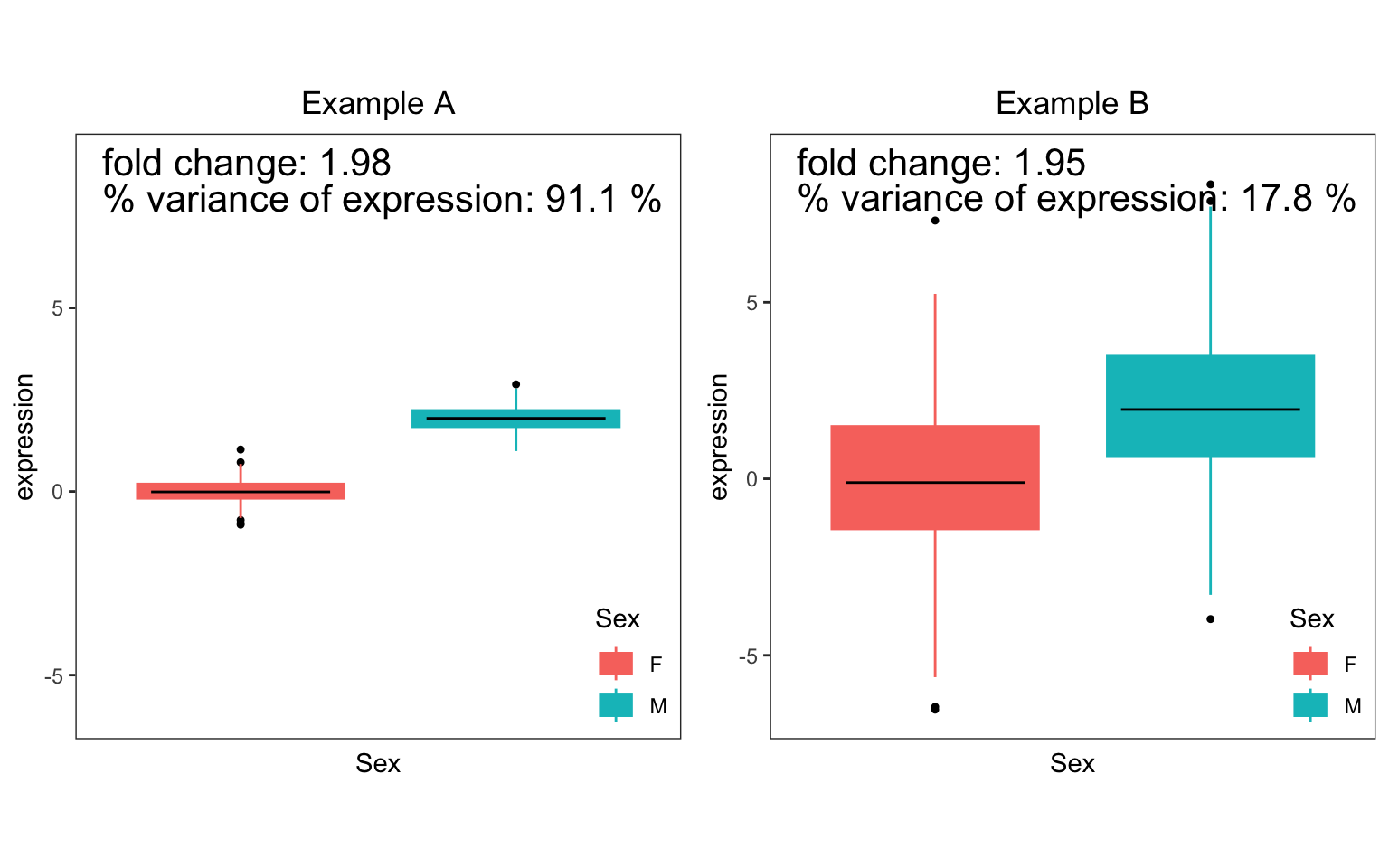
Differential expression (DE) is widely used to identify gene which show difference is expression between two subsets of the data (i.e. case versus controls). For a single gene, DE analysis measures the difference in mean expression between the two subsets. (Since expression is usually analyzed on a log scale, DE results are usually shown in terms of log fold changes between the two subsets ). In the Figure, consider two simulated examples of a gene whose expression differs between males and females. The mean expression in males is 0 and the mean expression in females is 2 in both cases. Therefore, the fold change is 2 in both cases.
However, the fraction of expression variation explained by sex is very different in these two examples. In example A, there is very little variation within each sex, so that variation between sexes is very high at 91.1%. Conversely, example B shows high variation within sexes, so that variation between sexes is only 17.8%.
The fact that the fold change or the fraction of variation is significantly different from 0 indicates differential expression between the two sexes. Yet these two statistics have different interpretations. The fold change from DE analysis tests a difference in means between two sexes. The fraction of variation explained compares the variation explained by sex to the total variation.
Thus the fraction of variation explained reported by
variancePartition reflects as different aspect of the data
not captured by DE analysis.
Modelling error in gene expression measurements
Uncertainty in the measurement of gene expression can be modeled with
precision weights and tests of differentially expression using
limma::voom() model this uncertainty directly with a
heteroskedastic linear regression (Law et al.
2014). variancePartition can use these precision
weights in a heteroskedastic linear mixed model implemented in
lme4 (Bates et al. 2015).
These precision weights are used seamlessly by calling
fitVarPartModel() or fitExtractVarPartModel()
on the output of limma::voom(). Otherwise the user can
specify the weights with the weightsMatrix parameter.
Session Info
## R version 4.5.1 (2025-06-13)
## Platform: aarch64-apple-darwin23.6.0
## Running under: macOS Sonoma 14.7.1
##
## Matrix products: default
## BLAS/LAPACK: /opt/homebrew/Cellar/openblas/0.3.30/lib/libopenblasp-r0.3.30.dylib; LAPACK version 3.12.0
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: America/New_York
## tzcode source: internal
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] dendextend_1.19.1 ballgown_2.40.0 DESeq2_1.48.2
## [4] SummarizedExperiment_1.38.1 Biobase_2.68.0 MatrixGenerics_1.20.0
## [7] matrixStats_1.5.0 GenomicRanges_1.60.0 GenomeInfoDb_1.44.3
## [10] IRanges_2.42.0 S4Vectors_0.48.0 BiocGenerics_0.54.1
## [13] generics_0.1.4 edgeR_4.6.3 lme4_1.1-38
## [16] Matrix_1.7-4 variancePartition_1.39.5 BiocParallel_1.42.2
## [19] limma_3.64.3 ggplot2_4.0.1
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 jsonlite_2.0.0 magrittr_2.0.4
## [4] farver_2.1.2 nloptr_2.2.1 rmarkdown_2.30
## [7] fs_1.6.6 BiocIO_1.18.0 ragg_1.5.0
## [10] vctrs_0.6.5 memoise_2.0.1 minqa_1.2.8
## [13] Rsamtools_2.24.1 RCurl_1.98-1.17 htmltools_0.5.8.1
## [16] S4Arrays_1.8.1 curl_7.0.0 broom_1.0.10
## [19] SparseArray_1.8.1 sass_0.4.10 KernSmooth_2.23-26
## [22] bslib_0.9.0 htmlwidgets_1.6.4 desc_1.4.3
## [25] pbkrtest_0.5.5 plyr_1.8.9 cachem_1.1.0
## [28] GenomicAlignments_1.44.0 lifecycle_1.0.4 iterators_1.0.14
## [31] pkgconfig_2.0.3 R6_2.6.1 fastmap_1.2.0
## [34] GenomeInfoDbData_1.2.14 rbibutils_2.4 digest_0.6.39
## [37] numDeriv_2016.8-1.1 AnnotationDbi_1.70.0 textshaping_1.0.4
## [40] RSQLite_2.4.4 labeling_0.4.3 mgcv_1.9-4
## [43] httr_1.4.7 abind_1.4-8 compiler_4.5.1
## [46] bit64_4.6.0-1 aod_1.3.3 withr_3.0.2
## [49] S7_0.2.1 backports_1.5.0 viridis_0.6.5
## [52] DBI_1.2.3 gplots_3.2.0 MASS_7.3-65
## [55] DelayedArray_0.34.1 rjson_0.2.23 corpcor_1.6.10
## [58] gtools_3.9.5 caTools_1.18.3 tools_4.5.1
## [61] remaCor_0.0.20 glue_1.8.0 restfulr_0.0.16
## [64] nlme_3.1-168 grid_4.5.1 reshape2_1.4.5
## [67] sva_3.56.0 gtable_0.3.6 tidyr_1.3.1
## [70] XVector_0.48.0 pillar_1.11.1 stringr_1.6.0
## [73] genefilter_1.90.0 splines_4.5.1 dplyr_1.1.4
## [76] lattice_0.22-7 survival_3.8-3 rtracklayer_1.68.0
## [79] bit_4.6.0 annotate_1.86.1 tidyselect_1.2.1
## [82] locfit_1.5-9.12 Biostrings_2.76.0 knitr_1.50
## [85] gridExtra_2.3 reformulas_0.4.2 RhpcBLASctl_0.23-42
## [88] xfun_0.54 statmod_1.5.1 stringi_1.8.7
## [91] UCSC.utils_1.4.0 yaml_2.3.10 boot_1.3-32
## [94] evaluate_1.0.5 codetools_0.2-20 tibble_3.3.0
## [97] cli_3.6.5 xtable_1.8-4 systemfonts_1.3.1
## [100] Rdpack_2.6.4 jquerylib_0.1.4 dichromat_2.0-0.1
## [103] Rcpp_1.1.0 EnvStats_3.1.0 png_0.1-8
## [106] XML_3.99-0.20 parallel_4.5.1 pkgdown_2.2.0
## [109] blob_1.2.4 bitops_1.0-9 viridisLite_0.4.2
## [112] mvtnorm_1.3-3 lmerTest_3.1-3 scales_1.4.0
## [115] purrr_1.2.0 crayon_1.5.3 fANCOVA_0.6-1
## [118] rlang_1.1.6 cowplot_1.2.0 KEGGREST_1.48.1